IGV可视化BAM文件帮助理解结构变异
使用二代测序数据可以检测结构变异,其主要利用insert size与建库时insert size的差异,以及pair-end orientation的异常来检测结构变异。
Insert size
二代测序,需要先将DNA打断成fragment length长的片段,然后加上测序接头,adapter等序列。insert size通常指文库长度,也就是两个reads起点和终点间的长度。
5' 3'
.....................................................
--------> <--------
read1 read2
-----------------
inner distance
gap size
-----------------------------------
insert size
-----------------------------------------------------
fragment length
上图分别标注了
- inner distance(gap size):两个reads之间的长度
- insert size:read1起点到read2终点的距离
- fragment length:insert size + adapter+index序列的长度
两个read重叠固然会提升该区域测序的准确度,但是这样测序的范围就更小了,如果我们使得两个read隔开,因为是随机打断的,所以中间那段gap size在其他read pair中会出现,这样中间的也被覆盖了,并且测得范围也更大了。
Pair orientation
与上图read1, read2对应,我们在测序时进行双端测序,那么假设read1方向是从5’—>3’ , 那么read2方向应为3’—>5’,正常情况下都是上述情况,为LR,但是当出现Inversion等变异时候,Pair orientation可能会发生改变,出现LL,RR,RL等情况。
Read1 Read2
-------> gap size <--------
异常检测
通过insert-size异常可以检测
- insertion (INS 插入)
- deletion (DEL 删除)
通过pair-end orientation异常可以检测
- inversion (INV 倒位)
- inverted duplication (倒位重复)
- Tandem duplication (DUP 串联重复)
- Translocation on the same chromosome (TRA 易位)
Deletion对应Bam文件在IGV中可视化的理解
首先我们考虑,如果样本基因组中某区域发生了DEL会产生什么现象
参考基因组
A B
---------------------------------------
在样本基因组中,假设A--B序列缺失,那么会有一对pair-end,read1在A的左边,read2在B右边,这一对reads比对到参考基因组的时候,产生的insert size将大于对样本建库时的insert size,因为在样本中他们已经是紧邻着的了,但是在参考基因组中他们中间相隔了一个AB序列.
比对到参考基因组
A B
-------------|··········|--------------
------------- 直接相连 --------------
----> <----
read1 read2
------------------------
样本中的reads-pair比对到
参考基因组的insert size
样本基因组比该片段要比上图的参考基因组短一点,
---------------------------
----> <----
read1 read2
-------------
insert size
很明显样本中的reads-pair比对到参考基因组产生的insert size大于该样本建库时的insert size或者说预期的insert size
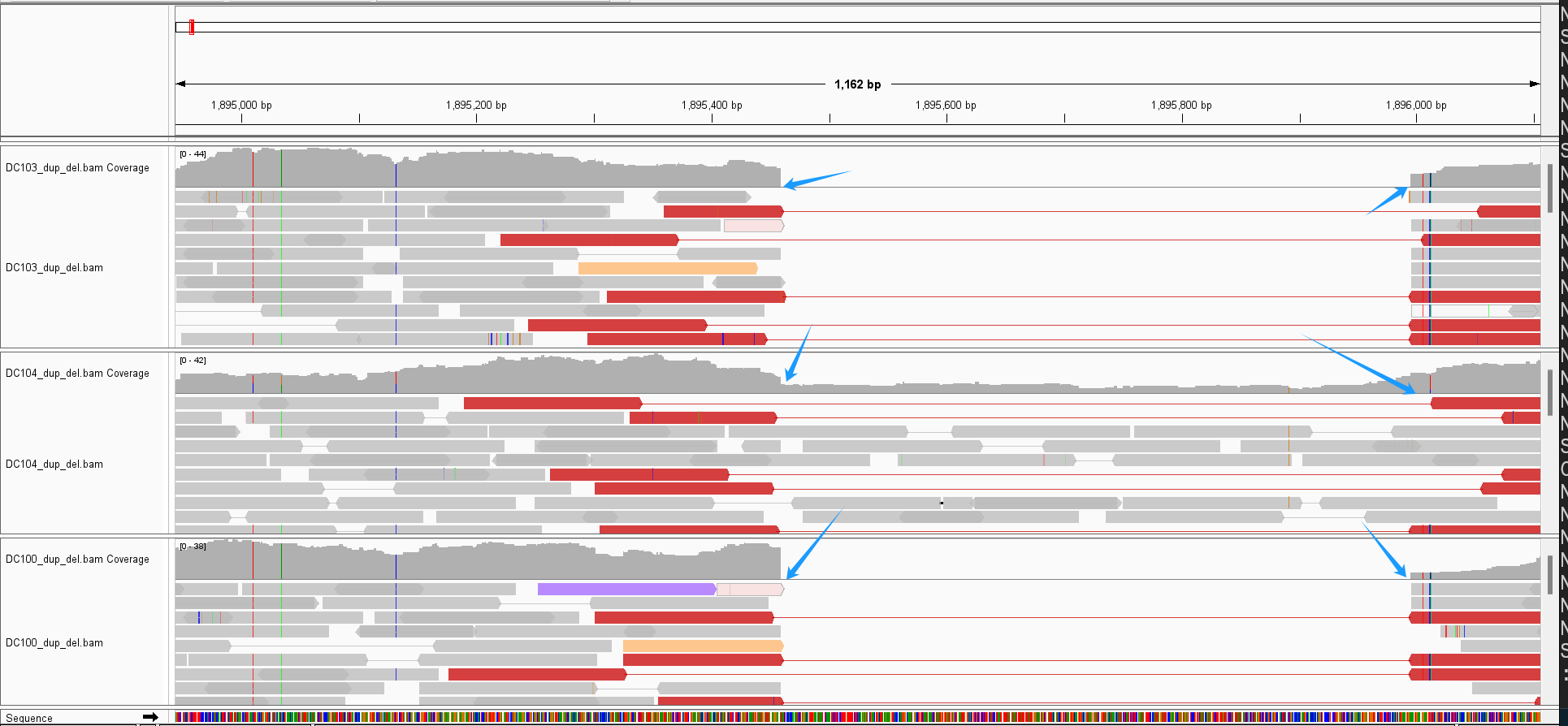
下面是一个DEL的例子,接下来看BAM文件在IGV中的展示
| CHROM | POS | ID | REF | ALT | QUAL | FILTER | INFO |
|---|---|---|---|---|---|---|---|
| NC_037328.1 | 1895458 | 9784 | N | <DEL> | 14942 | PASS | CIEND=0,0;CIPOS=-2,0;CHR2=NC_037328.1;END=1895994;SVLEN=-536;········ |
-
注意上图中蓝色箭头,在蓝色箭头处reads覆盖度发生了明显变化,再看下面红色的reads,这些就是insert size出现异常的reads
- 红色表示,比对到参考基因组的insert size大于建库时预期的insert size,也就是上文提到的DEL会出现的情况
- 中间样本除了红色reads,还有正常的Reads,覆盖了该区域,说明该样本只是部分变异,可以认为其一条染色体发生了该DEL,但是另一条并没有发生

如上图,随即点开一个标记为红色的read-pair,可以看到其insert size为844,但是建库时为300-500bp,显然这个insert size大于建库的insert size,那么判断发生了缺失
Insertion在bam文件中体现
同上面的DEL一样, 首先考虑,如果样本基因组中产生了INS会出现什么现象
|
------------------------------- 参考基因组
--------------······--------------- 样本基因组,中间部分发生了插入
------> <------
read1 read2 样本基因组建库测序产生
----------------------
insert size
但是我们要比对到的是参考基因组,由于参考基因组中没有中间插入的一段,所以将会造成以下结果
|
------------------------------- 参考基因组
------> <------
read1 read2
---------------
比对产生insert size
很显然,来自样本中的reads比对到参考基因组时,比对产生的insert size明显小于建库时的预期,推断此处发生了插入
下面是一个例子
vcf记录
| CHROM | POS | ID | REF | ALT | QUAL | FILTER | INFO |
|---|---|---|---|---|---|---|---|
| NC_037329.1 | 120047817 | NC_037329.1:120047817:IG | T | TGAACCCACATGTGTGATAGATGGTACA | 8962 | PASS | ABHet=0.542;ABHom=0.9893;AC=46;AF=0.4259;··· |
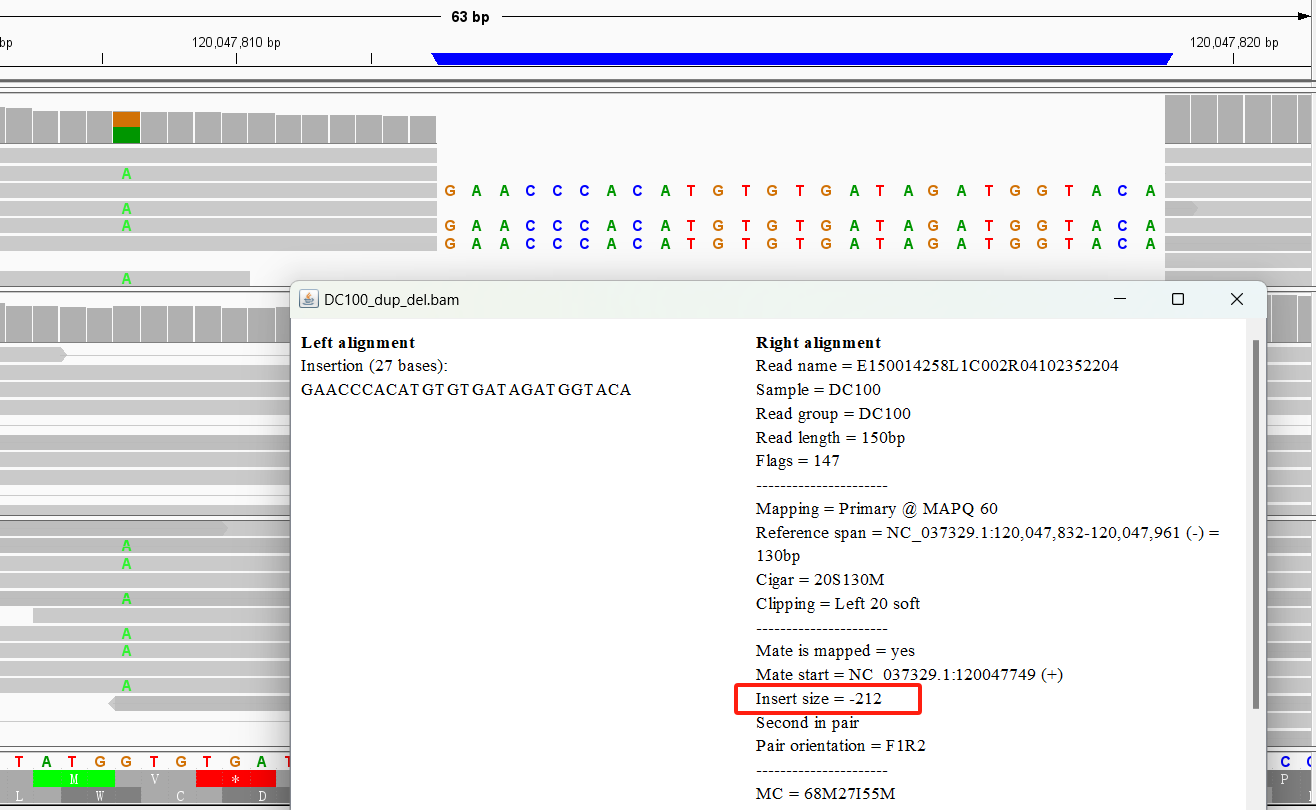
图中表示了一个27bp的插入,序列如图,并且能观察到该区域比对产生的insert size为212。显然,建库时我们预期为300-500bp显然更小,说明上述比对insert size < 建库insert szie时可能显示有插入发生,是合理的
** !! 注意:** *这种方式能检测插入的长度是有限的,当建库测序的inesrt size长度不足以覆盖整个插入片段以及两条reads时,那么read2将会有一部分在插入序列里面,此时比对,read2将会无法比对上,或者部分比对,此时没办法判断是因为read质量低还是插入或者其他情况造成的。*Inversion-倒位
倒位,指一个片段的DNA在原位置方向发生颠倒,下面看一下如果发生了倒位,比对到参考基因组将会发生什么事情
A B
--------------------------------------------- 参考基因组
B A
--------------------------------------------- 样本基因组
在样本基因组中,AB段序列与参考基因组相比发生了颠倒,但是我们不清楚,正常建库测序,假设我们在样本基因组中有以下测序片段
B A
--------------------------------------------- 样本基因组
------> <------
readB-1 readB-2
------> <------
readA-1 readA-2
现在将上述测序reads比对到参考基因组,将会发生什么呢?
A B
--------------------------------------------- 参考基因组
------> <------
readB-1 readA-1
------> <------
readB-2 readA-2
可以看到,在AB序列内部的reads,比对的方向居然和其配对的reads方向一致,也就是出现了LL,RR类型,指示可能发生了倒位。
Invert Duplication-倒位重复
倒位重复:指一小段序列在基因组上另一个位置发生了倒位并且插入进该位置
A B
------------------------------------ 参考基因组
A B B A
----------------------------------------------------- 样本基因组
在样本基因组中AB片段在另一个位置发生了倒位重复,我们对样本基因组进行建库测序
A B B' A'
------------------------------------------------------ 样本基因组
---->···<----
readA1 readA2
---->···<---- ---->···<----
readB1 readB2 readB'1 readB'2
---->···<----
readA'1 readA'2
将上述reads比对到参考基因组,将会发生什么呢?
A B
-------------------------------------------------- 参考基因组
---->···<----
readA1 readA2
---->···<---- ----> <----
readB1 readB2 readB'1 readA'2 ---->
readB'2
<----
readA'1
因为在样本基因组中有两段这个区域,所以比对后AB段的coverage(覆盖度)应该会有明显上升,IGV官方文档说会有大量的左右read重叠?有待考证···
Tandem duplication-串联重复
串联重复: 在一段序列后面紧跟着重复该片段
A B
----------------------------------- 参考基因组
A B
------------------------------------------------------------ 样本基因组
A B
--->···<---
read1 read2
上述样本基因组发生了AB序列串联重复,对其进行建库测序,再比对到参考基因组
A B
----------------------------------- 参考基因组
<--- --->
read2 read1
很显然read2本来应该再read1下游位置,但是跑到了上游,但是方向未变
IGV文档中没提到,这个位置的coverage应该也会上升
Translocation on the same chromosome-易位
同染色体易位: 染色体上的某一段序列,从原位置移动到该染色体的另一位置
A B
------------------------------------------------------------- 参考基因组
A B
------------------------------------------------------------- 样本基因组
---->···<----
read1 read2
上述样本基因组出现了同染色体易位变异,对其进行建库测序,再比对到参考基因组
A B
------------------------------------------------------------- 参考基因组
<---- ---->
read2 read1
可以观察到,测序产生reads比对到参考基因组后read1正常比对到原来的位置,但是配对的read2比对到了AB区间内,但是检测易位真是通过这个吗?
@TODO:研究一下calling sv的原理
End
以上内容来自本人对IGV中可视化bam文件部分的理解,因为似乎没有在互联网上找到相关的信息,直接从reads角度来看SV有助于更好理解结构变异的calling过程!