使用SV完成主成分分析
主成分分析(Principal component analysis)是一种降维的方法, 将高维特征映射到低维空间,也就是如果一个记录有很多特征,那么我们需要找到一组更少的特征来表示这众多的特征,这些更少的特征就称为主成分。
在群体基因组中,我们经常使用PCA来查看群体间的关系,以及群体内分层的现象等,大部分时间都是使用SNP来进行分析,也就是各群体间共有的SNP位点,对他们的基因型进行主成分分析。
本文将使用结构变异(Structural variation)进行主成分分析。
数据准备
在进行主成分分析之前,我们需要准备一个VCF文件,其中包含每个变异的相关信息,以及每个样本的基因型信息。
这里有个小坑,我的结构变异是分群体进行calling的,需要先得到群体间的公共位点才行,但是结构变异的断点本身就是不准确的,所以这一步还是很困难的。
我的处理步骤如下:
- 提取每个群体的位点
对每个群体提取所有变异位点,包含染色体号和变异起始位置
bcftools query -f '%CHROM\t%POS\n' your_vcf_file_1 | sort -V -k1,1 -k2,2n > site1.list
bcftools query -f '%CHROM\t%POS\n' your_vcf_file_2 | sort -V -k1,1 -k2,2n > site2.list
bcftools query -f '%CHROM\t%POS\n' your_vcf_file_3 | sort -V -k1,1 -k2,2n > site3.list
注意:上面命令中sort -V是按自然顺序排序,如果你文件中的染色体名是有英文字母的,如NCBI的NCxxxxx那么可以使用,如果你的染色体名字直接是数字那么就不需要加该选项
- 计算群体的共有位点
awk 'NR==FNR {pos[$0]; next} ($0 in pos)' site1.list site2.list site3.list > common.list
- 提取每个群体共有位点的变异
bcftools view -T common.list -O z -o population_1.vcf.gz vcf_1
bcftools view -T common.list -O z -o population_2.vcf.gz vcf_2
bcftools view -T common.list -O z -o population_3.vcf.gz vcf_3
- 使用bcftools进行合并(同时使用bcftools计算出AF,MAF,MISSING RATE)
bcftools merge population_1.vcf.gz population_2.vcf.gz population_3.vcf.gz -O z -o common_var.vcf.gz
bcftools +fill-tags common_var.vcf.gz -- -t AF | bcftools +fill-tags -- -t MAF | bcftools +fill-tags -O z -o common_var_with_info.vcf.gz -- -t F_MISSING
- 将vcf文件中的ALT字段全部替换成PLINK可接受的输入
# 此处假设vcf文件中ALT字段为<xxxxx>类型
# 使用sed替换
bcftools view common_var_with_info.vcf.gz | sed -E '/^#/! s/<[^>]+>/SV/g' > common_var_with_info.vcf
bgzip common_var_with_info.vcf
tabix common_var_with_info.vcf.gz
- 将vcf文件转换成PLINK的标准输入(使用PLINK2)
plink2 --vcf common_var_with_info.vcf.gz \
--geno 0.05 \ # 每个变异的missing rate < 5%
--maf 0.05 \ # 每个变异maf > 0.05
--hwe 1e-6 \ # 满足哈代温伯格平衡
--freq \ # 加载基因型频率
--allow-extra-chr \ # 由于我使用NCBI refseq的染色体ID,所以加上该选项,如果是数字染色体名则不需要
--make-bed \
--out plink/plink_input # 将结果存放到plink目录下,结果文件以plink_input为前缀
主成分分析及可视化
在将数据转换成PLINK标准输入后,使用PLINK继续进行主成分分析,并且使用R以及ggplot2包进行可视化。
- 计算主成分
plink2 --bfile plink_input --pca 10 -out pca_res
计算主成分后将会获得两个文件
- pca_res.eigenval (每个主成分的比例,无表头)
- pca_res.eigenvec(样本为行,主成分为列,有表头)
- 进行可视化
将上述两个文件,使用R打开,注意,我这里有三个群体,还需要手动在pca_res.eigenvec中加一下群体信息
eigenvec<-read.csv('pca_res.eigenvec', header=TRUE,sep = "\t")
eigenval<-read.table("pca_res.eigenval", header = FALSE)
# 去掉第一列并加上分组信息
library(dplyr)
eigenvec<-mutate(eigenvec[-1],group = substr(IID,1,2))
pca_data <- data.frame(
Sample = eigenvec[, 1], # 第一列样本信息
Group = factor(eigenvec$group),
PC1 = eigenvec[, 2], # 主成分1
PC2 = eigenvec[, 3] # 主成分2
)
# 计算方差解释比例
var_explained <- eigenval$V1 / sum(eigenval$V1) * 100
library('ggplot2')
# 绘制PCA图
ggplot(pca_data, aes(x = PC1, y = PC2, color = Group)) +
geom_point(size = 3, alpha = 0.7) +
labs(
x = paste0("PC1 (", round(var_explained[1], 2), "%)"),
y = paste0("PC2 (", round(var_explained[2], 2), "%)"),
) +
stat_ellipse(level = 0.95, linetype = 2, size = 0.5) + # 添加95%置信椭圆
theme_bw() +
theme(
legend.position = "top",
plot.title = element_text(hjust = 0.5)
)
然后就会得到如下PCA图
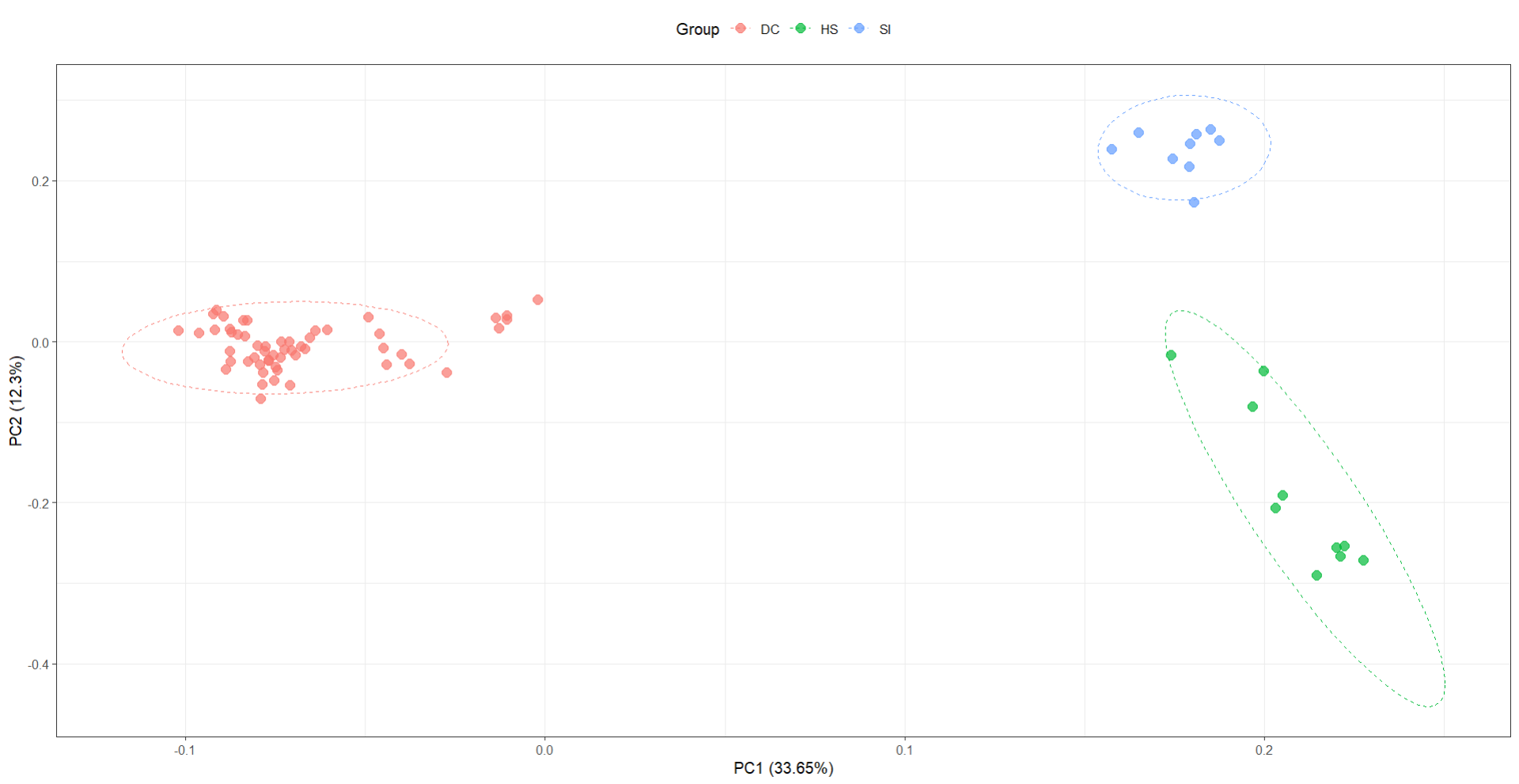
最后
使用结构变异来做PCA分析还是有很多小坑,特别是PLINK对SV支持不是很好,ALT列不能识别,需要手动替换