Pangenome学习:A gentle introduction to pangenomics
学习泛基因组的基本概念以及构建方法
Definition
- 一个物种或者一个群体存在的自然的变异,一个群体或者物种所有的基因组信息
- 使用生物信息学工具得到的泛基因组
Core genome
sequence shared by all individuals
Accessory / Shell genome
Sequence present in some but not all individuals
Dispensable / Cloud genome
Sequence found in only one individual
Type of pangenomes
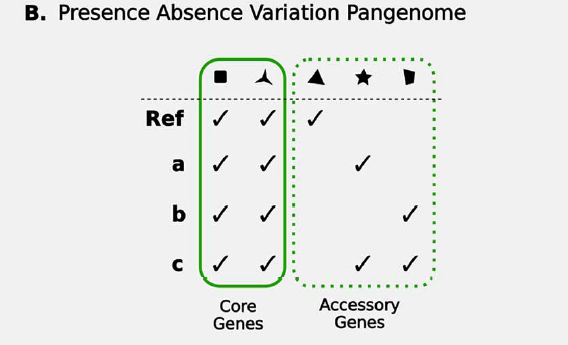
Prensence-absence variation pangenome (PAV)
关注基因的存在与否,不关注基因具体的位置,序列,以及多样性等
core genome : 群体中每个个体都存在的基因,或者物种间每个物种都存在的基因
accessory genome: 只在群体中部分个体存在的基因或者物种间部分物种存在的基因
Construction
homolog-based strategy
对每个样本进行de novo组装,组装后提取基因组中注释到的编码基因,并且在不同样本间根据基因序列相似性聚类,某个类别中如果包含所有的样本那么就认定为Core gene,否则认定为Accessory genes,通常用于原核生物,原核生物基因无内含子,比对,聚类成本较小。
map-to-pan strategy
将whole-genome sequencing reads比对到经过注释的具有代表性的泛基因组
- 测序深度最低10X
- 基因外显子至少5%的区域被至少1个reads覆盖,被认为基因存在的标志
softcore 当有一个样本中的Core gene没能被正确识别到的时候,那么整个Core gene合集可能就没有这个基因,为了防止这种情况,有些研究提出这个概念,允许在小部分样本中没识别到的基因为softcore
Representative sequence pangenome
使用尽可能少同源序列,并尽可能多的表示基因组多样性,通常由一个参考基因组以及其他非冗余参考序列组成(NRR, nonredundant reference), 其中NRR包含只在某一个成员中出现,但是不在reference中出现的

Construction
将在参考基因组中不存在的基因组区域作为额外contig来构建
- metagenome-like assembly of unaligned reads (低于10X的大样本测序数据首选)
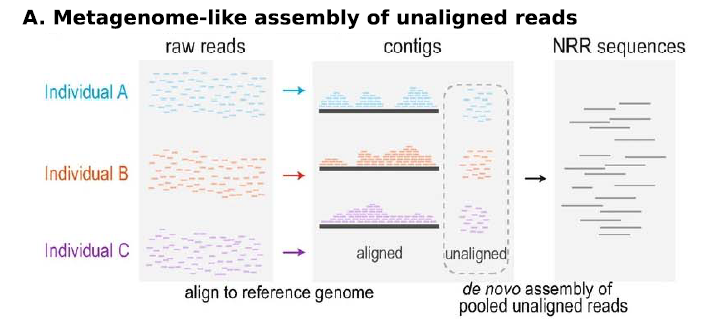
先将所有样本reads比对到参考基因组,没比对上的所有reads一起进行de novo assembly ,形成contig
- independent assembly of unaligned reads
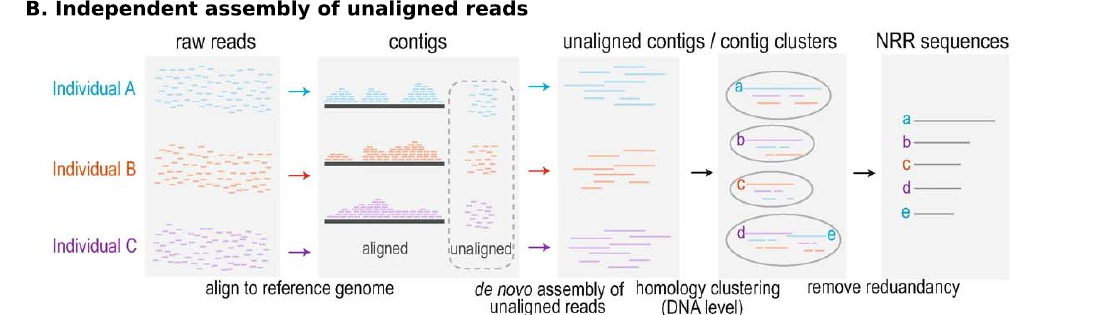
先将每个样本reads比对到参考基因组,没比对上的按样本进行de novo组装,将组装好的contig根据序列相似度进行聚类,从每个聚类中选出一个作为参考基因组的补充(需要至少10x的read coverage)
- iterative assembly of unaligned reads
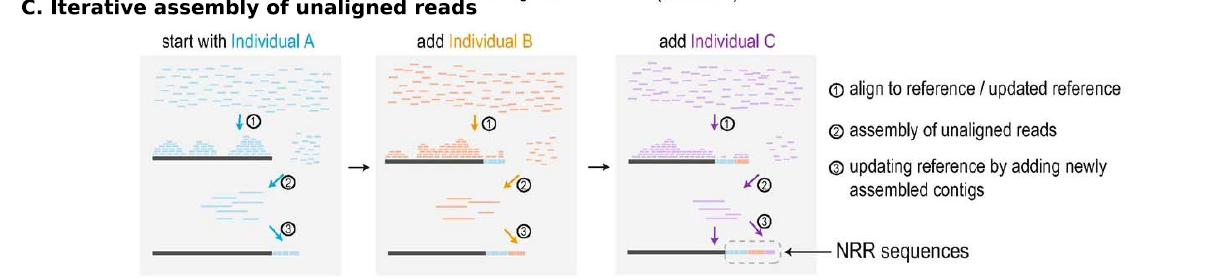
迭代式创建,单个样本先进行比对,然后将未比对的reads进行de novo assembly,用于扩充参考基因组,然后更新的参考基因组再用于其他样本。后续样本就先比对到更新的参考基因组,然后扩充参考基因组,一直迭代。
- independent whole-genome assembly
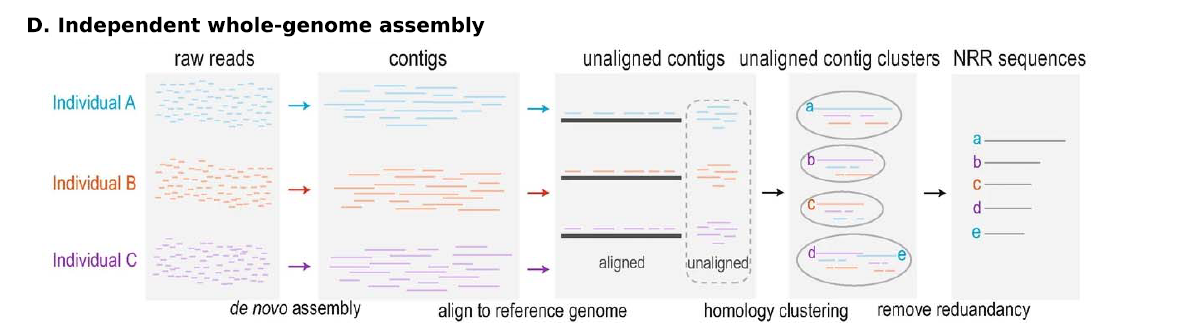
先将每个样本的reads进行de novo assembly至contig水平,然后将contig比对到参考基因组,所有样本未能比对至参考基因组的contigs进行聚类,从聚类结果中挑选每个类别中最长的序列用于扩充参考基因组
pangenome graph (graphic pangenome)
可以是面向序列的和面向基因的,展示群体中变异和基因的位置关系。
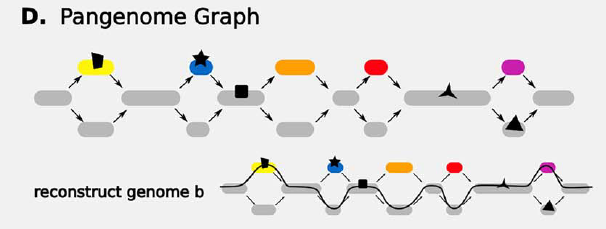
Construction
- Predetermined variants
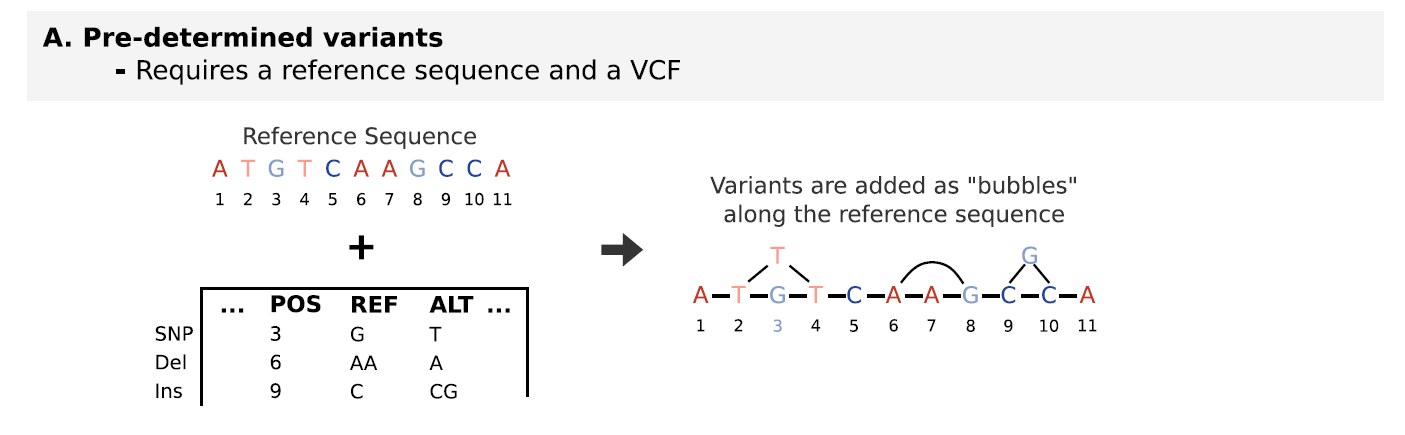
需要一个参考基因组,以及包含已有变异的VCF文件,后来新添加的变异作为分支节点添加到图上,形成一个有向无环图
- Multiple sequence alignment
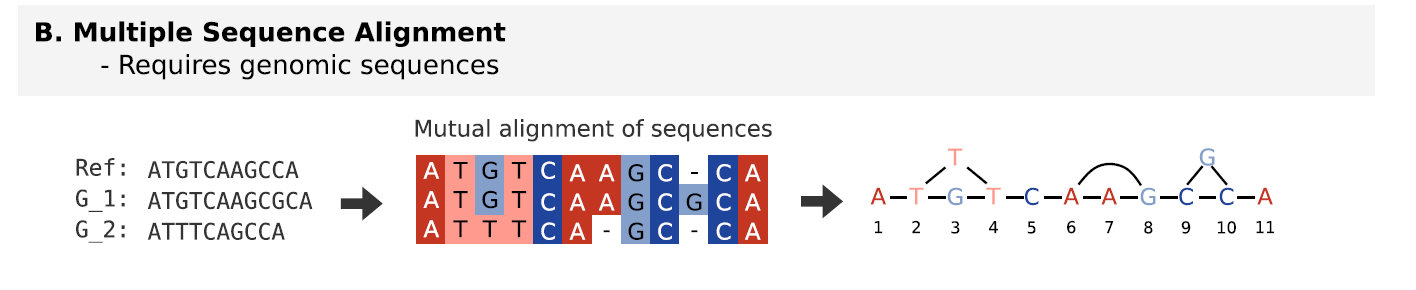
多序列比对,直接将序列两两比对进行图形基因组构建,这种方法需要大量的计算资源,但是对于定相的基因组十分有用(Phased genome)
- De Bruijn graphs
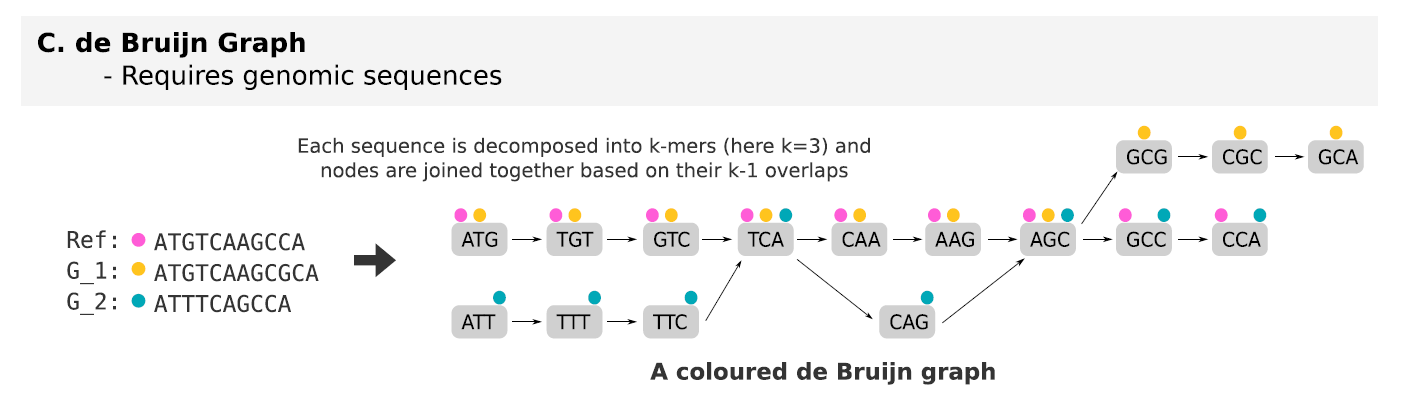
将reads分成k-mers,然后根据k-1(k为kmer的长度)的重叠来进行图构建,kmer为图节点
Applications
- 变异检测与分型(variants calling and genotyping)
- 单倍型推断 (Haplotype inference; phasing of assemblies)
Files and formats
| Linear | Pangenomic | |
|---|---|---|
| Reference genome | .fa | .gfa |
| Indexing | .bwt | .gbwt |
| Alignment | .bam / .paf | .gam / .gaf |
Reference
Chelsea A Matthews, Nathan S Watson-Haigh, Rachel A Burton, Anna E Sheppard, A gentle introduction to pangenomics, Briefings in Bioinformatics, Volume 25, Issue 6, November 2024, bbae588, https://doi.org/10.1093/bib/bbae588